Co to jest i jakie są przyczyny?
Choroba Wilsona jest schorzeniem uwarunkowanym genetycznie. W następstwie mutacji genu ATP7B, włączonego w regulację równowagi metabolizmu miedzi, dochodzi do nadmiernego gromadzenia tego metalu w organizmie.
Defekt genetyczny powoduje, że nadmiar miedzi wchłoniętej z przewodu pokarmowego nie jest wydalany przez wątrobę i gromadzi się w jej komórkach. Toksyczne oddziaływanie metalu prowadzi do zróżnicowanych form uszkodzenia wątroby.
Z czasem komórki wątroby tracą zdolność gromadzenia miedzi i następuje jej uwalnianie do krążenia. W tym okresie choroby następuje szkodliwe oddziaływanie miedzi na ośrodkowy układ nerwowy i nerki, a także stwierdzane jest odkładanie złogów w rogówce oczu. Zwiększenie ilości miedzi w nerkach wywołuje zwykle nikłe zmiany strukturalne i niewielkie funkcjonalne.
Jednoznacznie toksyczny wpływ miedzi zaznacza się natomiast w obrębie ośrodkowego układu nerwowego z uszkodzeniem jego struktur, martwicą komórek nerwowych i, wtórnie, powstawaniem jamistości tkanki nerwowej. W okresie uwalniania miedzi do krążenia występujące zaburzenia metaboliczne krwinek mogą prowadzić do przełomów hemolitycznych – nagłego rozpadu dużej liczby erytrocytów. Bardzo rzadko obserwuje się cechy uszkodzenia mięśnia serca, gruczołów wewnętrznego wydzielania, zmiany kostne i stan zapalny stawów.
Gromadzenie miedzi w chorobie Wilsona rozpoczyna się od momentu urodzenia, jednak manifestacja choroby rzadko występuje przed 6. rokiem życia, najczęściej objawiając się w okresie dojrzewania. W przypadku braku leczenia postępuje u wszystkich pacjentów.
Jak często występuje choroba Wilsona?
Zapadalność na chorbę Wilsona wynosi 1 na 30—100 tysięcy mieszkańców, częstość nosicielstwa nieprawidłowego genu warunkującego tę chorobę jest większa i wynosi około 1 na 90. Choroba Wilsona występuje w przypadku obecności 2 nieprawidłowych kopii genu, równocześnie odziedziczonych przez chorego od matki i ojca. Obecność tylko 1 kopii zmutowanego genu nie objawia się klinicznie z powodu dominacji genu prawidłowego – osoba taka jest zwykle bezobjawowym nosicielem. Lokalizacja genu na chromosomie niezwiązanym z płcią powoduje brak różnic częstości występowania schorzenia u kobiet i mężczyzn.
Jak się objawia choroba Wilsona?
Kliniczne objawy choroby Wilsona rzadko występują przed 6. rokiem życia, a najczęściej pojawiają się w okresie dojrzewania (aczkolwiek istnieją pojedyncze opisy przypadków choroby Wilsona rozpoznanych po 60. roku życia).
Postać wątrobowa choroby Wilsona może przypominać różne schorzenia tego narządu – od bezobjawowego podwyższenia enzymów wątrobowych i stłuszczenia, do ostrego i nadostrego zapalenia wątroby, a także marskości.
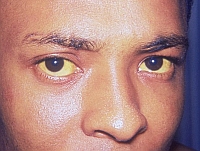
W młodszych grupach wiekowych uszkodzenie wątroby poprzedza o około dziesięć lat rozwój zmian w ośrodkowym układzie nerwowym. Jednak to dopiero zaburzenia neurologiczne lub psychiatryczne są zwykle pierwszymi objawami, na podstawie których rozpoznaje się chorobę Wilsona. W tym okresie stwierdzane jest już zaawansowane gromadzenie (spichrzanie) miedzi w organizmie manifestujące się, poza uszkodzeniem ośrodkowego układu nerwowego, odkładaniem złogów w rogówce w postaci pierścieni Kaysera-Fleischera (zob. fotografię) oraz, rzadziej, tzw. zaćmy słonecznikowej.
Objawy neurologiczne są podobne jak w chorobie Parkinsona:
- drżenia spoczynkowe i zamiarowe
- sztywność mięśni
- pląsawica (mimowolne skurcze mięśni lub grup mięśniowych, powodujące dziwaczne ruchy różnych okolic ciała i grymasy twarzy)
- ślinienie się
- utrudnione połykanie i upośledzenie mowy.
Poza bólem głowy zmiany czuciowe nie występują. Zaburzenia psychiczne u większości chorych łączą się z objawami neurologicznymi; występują w postaci schizofrenii, zaburzeń maniakalno-depresyjnych i nerwicy.
Objawami zmian w nerkach są krwinkomocz, białkomocz, rzadko - wapnica i kamica nerek oraz złożone zaburzenia czynności cewek nerkowych.
Zmiany w innych narządach i tkankach dotyczą gruczołów wydzielania wewnętrznego i pojawiają się rzadko. Są to: niedoczynność tarczycy i przytarczyc, zaburzenia miesiączkowania oraz powtarzające się poronienia w przypadku zajścia w ciążę. Możliwe są także zaburzenia kardiologiczne (pod postacią kardiomiopatii), uszkodzenia mięśni szkieletowych, układu kostnego oraz zapalenia stawów.
Co robić w razie wystąpienia objawów?
W razie wystąpienia objawów sugerujących chorobę Wilsona, należy zgłosić się do lekarza pierwszego kontaktu. Skieruje on pacjenta do lekarza specjalisty albo do szpitala.
Wskazaniem do diagnostyki, poza podejrzeniem choroby, jest także występowanie jej u osób blisko spokrewnionych (dziadkowie, rodzice, rodzeństwo, dzieci).
Jak lekarz ustala diagnozę?
Kliniczne podejrzenie choroby Wilsona wymaga wykonania badań laboratoryjnych oceniających metabolizm miedzi. Wskazaniem do testów jest także występowanie choroby Wilsona wśród członków najbliższej rodziny. Badania obejmują określenie stężenia ceruloplazminy w surowicy, stężenia miedzi w surowicy i miąższu wątroby, dobowego wydalania miedzi z moczem.
Analizę genetyczną mutacji genu ATP7B przeprowadza się za pomocą metod molekularnych, umożliwiających potwierdzenie oraz dokładne określenie rodzaju zmian genetycznych.
Ważnym badaniem jest ocena obecności złogów miedzi w rogówce oka w postaci pierścienia Kaysera-Fleischera. Badanie to powinno być wykonywane przez doświadczonego okulistę, z użyciem tzw. lampy szczelinowej. W przypadku marskości wątroby lub zaawansowanych zmian neurologicznych, brak powyższych zmian okulistycznych podważa rozpoznanie choroby Wilsona jako przyczyny obserwowanych uszkodzeń narządowych. W przypadku mniej zaawansowanych zmian wątroby, brak złogów miedzi w rogówce stwierdzany jest jedynie u około 15% chorych.
Podsumowując, rozpoznanie choroby Wilsona jest wysoce prawdopodobne, gdy:
- stężenie ceruloplazminy jest małe
- stwierdzana jest obecność pierścienia Kaysera-Fleischera ze zwiększonym wydalaniem miedzi z moczem
- zawartość miedzi w wątrobie przekracza 250 µg/g suchej masy.
Jakie są sposoby leczenia?
Leczenie choroby Wilsona polega na usuwaniu nadmiaru miedzi z organizmu. Dostępne metody terapii to zahamowanie wchłaniania, eliminacja miedzi z istniejących zasobów oraz, w niektórych okolicznościach, przeszczepienie wątroby.
Leczenie dietetyczne polega na eliminacji z diety produktów bogatych w miedź, takich jak:
- ryby
- orzechy
- podroby (wątroba)
- czekolada
- grzyby.
Z powodu możliwości zwiększonego stężenia miedzi w wodzie pochodzącej z niektórych źródeł oraz systemów wodociągowych, wskazana jest ocena zawartości miedzi i ewentualna zmiana rodzaju spożywanej wody. Wskazana jest także eliminacja sprzętów kuchennych, które bezpośrednio stykają się z żywnością i zawierają w swojej konstrukcji elementy miedziane. Ten rodzaj czynności profilaktycznych nie jest wystarczający i powinien być stosowany w łączności z leczeniem farmakologicznym.
Leczenie farmakologiczne pozostaje główną metodą terapii choroby Wilsona i dzieli się na 2 etapy. Pierwszy polega na obniżeniu stężenia miedzi w tkankach poniżej poziomu toksycznego, drugi – na utrzymywaniu jej niskiego stężenia przez terapię podtrzymującą. Leczenie powinno być wprowadzone bezpośrednio po ustaleniu rozpoznania, bez względu na występowanie lub brak objawów chorobowych. Terapia opiera się na stosowaniu dwóch leków: penicylaminy i, zastępczo, trietylenotetraminy.
Penicylamina pozostaje od 1956 roku standardem leczenia choroby Wilsona. Właściwości tych leków polegają na wiązaniu atomów miedzi, tworzeniu rozpuszczalnych w wodzie kompleksów i wydalaniu ich przez nerki. Kontynuowanie długotrwałej terapii skutecznie zmniejsza zasoby zgromadzonej w organizmie miedzi. Głównym problemem terapii są liczne i częste działania niepożądane, wymagające stałego nadzoru. W przypadku wystąpienia działań niepożądanych, konieczne jest przerwanie terapii i ponowne jej włączenie w momencie ustąpienia objawów – początkowo z użyciem małych, a następnie zwiększanych dawek. W przypadku wystąpienia ciężkich powikłań lub przeciwwskazań do podawania penicylaminy istnieje konieczność zmiany terapii z wprowadzeniem do leczenia trietylenotetraminy. Leczenie cynkiem ma znaczenie pomocnicze i umożliwia terapię mniejszymi dawkami leków podstawowych.
Uwaga!
Leczenie musi trwać nieprzerwanie przez całe życie chorego, gdyż tylko ten sposób zapewnia bezobjawowy przebieg choroby. Wymagane jest kontynuowanie terapii nawet w ciąży, a jej zakończenie jest możliwe jedynie w przypadku przeszczepienia wątroby.Czy możliwe jest całkowite wyleczenie?
Nieleczona choroba postępuje, rozwija się marskość wątroby, nasilają zaburzenia neurologiczne lub psychiczne. Wczesne rozpoznanie pozwala na rozpoczęcie leczenia, łagodzące objawy oraz zapobiegające powikłaniom i śmierci. Leczenie musi trwać nieprzerwanie przez całe życie chorego.
Co trzeba robić po zakończeniu leczenia?
Leczenie choroby Wilsona trwa całe życie, z wyjątkiem osób, które przebyły przeszczepienie wątroby. Po przeszczepieniu należy stosować się do zaleceń lekarzy chirurgów transplantologów, szczególnie odnośnie przyjmowania leków zapobiegających odrzuceniu przeszczepu.
Co robić, aby uniknąć zachorowania?
Nie można zapobiec wystąpieniu choroby Wilsona.